文献情報
- タイトル: Variational quantum simulation for periodic materials
- 著者:Nobuyuki Yoshida, Takeshi Sato, Yuya O. Nakagawa, Yu-ya Ohnishi, Wataru Mizukami
- 書誌情報(DOI):https://doi.org/10.1103/PhysRevResearch.4.013052
- 出版年:2022年1月
概要
物質の電子状態を求めることができれば、誘電率、バンドギャップ、光吸収特性など、さまざまな物性値を理論的に評価できる。電子状態を記述する基本方程式はシュレディンガー方程式であるが、多数の電子を含む実際の原子・分子・固体に対してこれを厳密に解くことは極めて困難である。そのため、実用的な電子状態計算では、波動関数や電子密度に対して何らかの近似を導入し、計算可能な形に定式化する必要がある。
経験的パラメータに頼らず、量子力学の基本法則に基づいて電子状態や全エネルギーを求める計算手法は、第一原理計算と呼ばれる。代表的な手法として、Hartree–Fock法、密度汎関数理論(Density Functional Theory: DFT)、Møller–Plesset摂動論(MP2法)、結合クラスター法(Coupled Cluster: CC法)、全配置間相互作用法(Full Configuration Interaction: Full CI法)などがある。これらの手法は、物質設計、材料開発、物性予測などの分野で広く用いられている。
近年では、量子コンピュータを用いた電子状態計算手法として、Variational Quantum Eigensolver(VQE)が注目されている。VQEは、量子コンピュータ上でパラメータ化された試行波動関数を生成し、そのエネルギー期待値を測定する一方で、古典コンピュータを用いてパラメータを最適化する量子–古典ハイブリッドアルゴリズムである。特に、古典計算で扱いが難しい強相関電子系に対して、有望な手法として研究が進められている。
本記事で取り上げる論文では、周期的な一次元水素鎖を対象として、強相関領域におけるポテンシャルエネルギー曲線の計算を行っている。古典的な近似手法では精度が低下しやすい領域において、VQEに基づくUCCSD型の手法がFull CIに近い精度を示すことが報告されている。本記事では、その計算手法の概要と論文の結果について解説する。
背景
量子多体系の電子状態を求める際には、対象とする系の情報を含んだハミルトニアンを定義し、その固有値問題を解くことでエネルギー固有値と対応する固有状態を得る、というアプローチをとる。
時間に依存しないシュレディンガー方程式
$$\hat{H}\ket{\psi_n}=E_n\ket{\psi_n}$$
と表される。ここで、$\hat{H}$ はハミルトニアン、$\ket{\psi_n}$ は固有状態、$E_n$ は対応するエネルギー固有値である。
しかし、多数の電子を含む系では、波動関数は多数の粒子座標に依存する高次元の関数となる。電子数を N とすると、空間座標だけでも 3N 次元の自由度をもつため、シュレディンガー方程式を厳密に解くことは一般に困難である。したがって、実際の電子状態計算では、ハミルトニアン、波動関数、電子密度、電子相関などに対して近似を導入し、計算可能な形に定式化する必要がある。
第一原理計算とは、経験的パラメータに頼らず、量子力学の基本法則に基づいて電子状態や全エネルギーを求める計算手法の総称である。多電子系のシュレディンガー方程式を厳密に解くことは困難であるため、実際には波動関数や電子密度に対して近似を導入し、コンピュータで計算可能な形に定式化する。
第一原理計算によって得られる全エネルギーや電子状態の情報を用いることで、原子構造の安定性、構造最適化、バンド構造、スピン状態、誘電率、光吸収係数など、さまざまな物性を評価できる。そのため、第一原理計算は物質設計、材料開発、物性予測などの分野で広く用いられている[1]。
古典コンピュータ上で用いられてきた代表的な電子状態計算手法には、Hartree–Fock法(HF法)[2,3]、Møller–Plesset摂動論(MP2法)[18]、結合クラスター法(CC法)[19]、全配置間相互作用法(Full CI法)[4]、密度汎関数理論(DFT)[5,6]などがある。
HF法は計算コストを比較的低く抑えられる一方で、電子を独立に運動する粒子として扱う平均場近似に基づくため、電子同士が互いの位置を避けながら運動する効果を十分に記述できない。Full CI法は、与えられた基底関数から構成できるすべての電子配置(スレーター行列式)を用いて波動関数を展開するため、その基底関数空間内でシュレーディンガー方程式を厳密に解いた結果と一致する高精度な手法であるが、計算コストが電子数や軌道数に対して急激に増大する。DFTは、電子密度を基本変数として扱うことで比較的低い計算コストと実用的な精度を両立できるため、固体材料を対象とする第一原理計算で広く用いられている。
近年では、量子コンピュータを用いた電子状態計算アルゴリズムとして、VQE(Variational Quantum Eigensolver)[6]が注目されている。VQEは、パラメータ化された試行波動関数を量子コンピュータ上で生成し、そのエネルギー期待値を測定し、パラメータを最適化を古典コンピュータ上で行う量子–古典ハイブリッド手法である。
VQEは、変分原理に基づいて基底状態エネルギーを近似的に求める手法であり、特に電子間の相互作用が強く、各電子を平均的な場の中で独立に運動する粒子として近似することが難しい強相関電子系のように、古典計算で扱いが難しい問題に対して有望であると考えられている。また、量子位相推定法のような深い量子回路を必要とする手法とは異なり、パラメータ化量子回路の深さをハードウェア性能に応じて調整できるため、比較的浅い量子回路でも実行可能である。この特徴から、誤り耐性量子コンピュータが実現する以前のNISQデバイスに適したアルゴリズムの一つとして注目されている[7,8]。
VQEでは、量子コンピュータ上で生成する試行波動関数の形をあらかじめ決めておき、その中に含まれるパラメータを最適化する。この試行波動関数の構成方法のことをansatzと呼ぶ。代表的なansatzとして、量子化学計算でよく用いられるUnitary Coupled Cluster(UCC)法[9,10]や、量子デバイス上で実装しやすいhardware-efficient ansatz[11]がある。
本記事では、VQEで用いられる代表的なansatzの一つであるUCC法、特に単一励起および二重励起を取り入れたUCCSD法について説明する。
手法
VQEの理論的基盤は変分原理である。変分原理によれば、任意の規格化された試行波動関数 $\ket{\psi(\theta)}$ に対するエネルギー期待値$E(\theta)=\braket{\psi(\theta)|\hat{H}|\psi(\theta)}$は、真の基底状態エネルギー $E_0$以上となる。すなわち、
$$E(\theta) \geq E_0$$
が成り立つ。したがって、パラメータ $\theta$を変化させて $E(\theta)$を最小化することで、基底状態エネルギーを近似的に求めることができる。
本記事では、周期系に対するVQE計算の流れを説明し、その中で用いられるUCCSD型ansatzとハミルトニアンの測定方法について述べる。
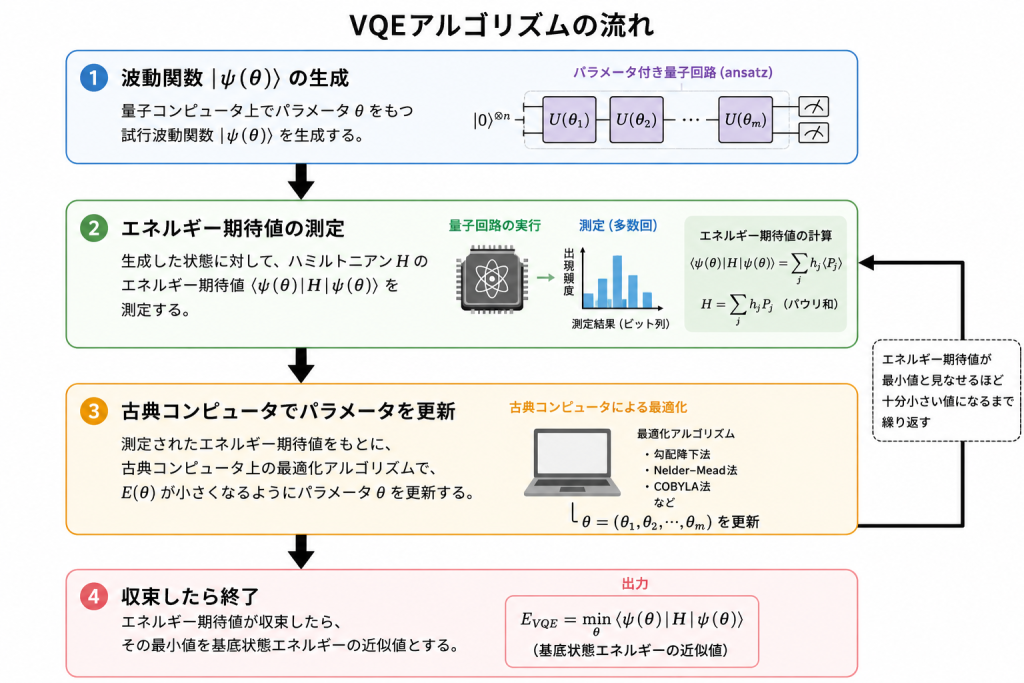
VQEのアルゴリズムについて
VQEの基本的な流れは次のようにまとめられる。
- 量子コンピュータ上で、パラメータ $\theta$ をもつ試行波動関数 $\ket{\psi(\theta)}$ を生成する。
- 生成した状態に対して、ハミルトニアン $\hat{H}$ のエネルギー期待値$E(\theta)=\braket{\psi(\theta)|\hat{H}|\psi(\theta)}$を測定する。
- 測定されたエネルギー期待値を古典コンピュータに渡し、最適化アルゴリズムを用いて $E(\theta)$ が小さくなるようにパラメータ $\theta$ を更新する。
- エネルギー期待値が収束するまで、状態生成、測定、パラメータ更新を繰り返す。得られた最小値を基底状態エネルギーの近似値とする。
1. ansatzの構成
VQEでは、試行波動関数 $\ket{\psi(\theta)}$ の形をあらかじめ決め、その中に含まれるパラメータ $\theta$ を最適化する。電子状態計算でよく用いられるansatzの一つに、Unitary Coupled Cluster(UCC)法がある。
UCC法は、古典的な結合クラスター(Coupled Cluster: CC)法の考え方を量子コンピュータ上で扱えるように拡張した手法である。通常のCC法では、参照波動関数 $\ket{\Phi_0}$ に対して指数型演算子 $e^T$ を作用させる。一方、UCC法では、反エルミート演算子 $T-T^\dagger$ を用いて、
$$\ket{\Psi_{\mathrm{UCC}}}=e^{T-T^\dagger}\ket{\Phi_0}$$
と試行波動関数を構成する。ここで、$\ket{\Phi_0}$ は参照波動関数であり、通常はHartree–Fock波動関数が用いられる。また、$T$ は励起演算子、$T^\dagger$ はそのエルミート共役である。
通常のCC法で用いられる $e^T$ は一般にユニタリー演算子ではないため、そのまま量子回路として実装することは難しい。一方、UCC法で用いられる $T-T^\dagger$ は反エルミート演算子であるため、$e^{T-T^\dagger}$ はユニタリー演算子となり、量子回路として実装しやすい。
UCCSD法では、励起演算子 $T$ を励起次数に関してクラスター展開し、単一励起項 $T_1$ と二重励起項 $T_2$ までで打ち切る。すなわち、
$$T \simeq T_1+T_2$$
と近似する。ここで、
$$T_1=\sum_{ia} t_i^a a_a^\dagger a_i$$
$$T_2=\sum_{ijab} t_{ij}^{ab} a_a^\dagger a_b^\dagger a_j a_i$$
である。$i,j$ はHartree–Fock参照状態における占有軌道、$a,b$ は非占有軌道を表す。また、$t_i^a$ および $t_{ij}^{ab}$ は励起振幅であり、VQEではこれらをパラメータとして最適化する。ここで、Hartree–Fock参照状態$\ket{\Phi_0}$ とはHartree–Fock法によって得られる近似基底状態であり、UCCSDではこの状態を基準として電子励起を導入する。
したがって、UCCSD ansatzは、
$$\ket{\Psi_{\mathrm{UCCSD}}}=e^{T_1+T_2-T_1^\dagger-T_2^\dagger}\ket{\Phi_0}$$
と表される。実際の量子回路実装では、この指数演算子を直接実装することは困難であるため、演算子を実装可能な量子ゲート列へ分解する近似手法であるTrotter分解などによって、量子ゲート列として近似的に表現する。
電子の生成消滅演算子を量子ビット上で扱うためには、フェルミオン演算子をパウリ演算子に変換できれば十分である。その代表的な方法がJordan–Wigner変換[12]である。Jordan–Wigner変換を用いると、生成演算子 $a_j^\dagger$および消滅演算子 $a_j$ は、
$$a_j^\dagger=\left(\prod_{k=0}^{j-1} Z_k\right)\frac{X_j-iY_j}{2}$$
$$a_j=\left(\prod_{k=0}^{j-1} Z_k\right)\frac{X_j+iY_j}{2}$$
と表される。ここで、$X_j、Y_j、Z_j$ は $j$ 番目の量子ビットに作用するパウリ演算子である。
この変換により、UCCSD ansatzに含まれる生成消滅演算子をパウリ演算子の積として表すことができる。したがって、UCCSD波動関数は、パラメータ付き量子ゲート列として量子コンピュータ上に実装できる。
2. エネルギー期待値を測定する
VQEでは、量子コンピュータ上で生成した試行波動関数 $\ket{\psi(\theta)}$ に対して、ハミルトニアン $\hat{H}$ の期待値
$$E(\theta)=\braket{\psi(\theta)|\hat{H}|\psi(\theta)}$$
を測定する。
周期境界条件をもつ固体電子系のフェルミオンハミルトニアンは、第二量子化表示を用いて一般に
$$
\hat{H}=
\sum_{p,q}\sum_{\boldsymbol{k}}t_{pq}^{\boldsymbol{k}}\hat{c}_{p\boldsymbol{k}}^{\dagger}\hat{c}_{q\boldsymbol{k}}
+\sum_{p,q,r,s}\sum_{\boldsymbol{k}_{p},\boldsymbol{k}_{q},\boldsymbol{k}_{r},\boldsymbol{k}_{s}}v_{pqrs}^{\boldsymbol{k}_{p}\boldsymbol{k}_{q}\boldsymbol{k}_{r}\boldsymbol{k}_{s}}\hat{c}_{p\boldsymbol{k}_{p}}^{\dagger}\hat{c}_{q\boldsymbol{k}_{q}}^{\dagger}\hat{c}_{r\boldsymbol{k}_{r}}\hat{c}_{s\boldsymbol{k}_{s}}$$
のように表される。ここで、$\hat{c}_{p\boldsymbol{k}}^{\dagger}$ および $\hat{c}_{p\boldsymbol{k}}$ は、それぞれ結晶運動量 $\boldsymbol{k}$ をもつ $p$ 番目の結晶軌道の生成演算子および消滅演算子である。また、$t_{pq}^{\boldsymbol{k}}$ は一電子積分、$v_{pqrs}^{\boldsymbol{k}_{p}\boldsymbol{k}_{q}\boldsymbol{k}_{r}\boldsymbol{k}_{s}}$ は二電子積分を表す。
これらの積分値は、VQEを実行する前に古典コンピュータ上で計算される。得られたフェルミオンハミルトニアンにJordan–Wigner変換などを適用すると、量子ビットハミルトニアン
$$\hat{H}=\sum_\ell h_\ell P_\ell$$
として表すことができる。ここで、$h_\ell$ は古典計算から得られる係数、$P_\ell$ はパウリ演算子の積である。
したがって、エネルギー期待値は
$$E(\theta)=\sum_\ell h_\ell\braket{\psi(\theta)|P_\ell|\psi(\theta)}$$
と分解できる。量子コンピュータでは各パウリ演算子 $P_\ell$ の期待値を測定し、それに係数 $h_\ell$ を掛け合わせて足し合わせることで、エネルギー期待値 $E(\theta)$ を評価する。
3. 古典コンピュータでパラメータを最適化する
量子コンピュータで測定されたエネルギー期待値 $E(\theta)$ は、古典コンピュータ上の最適化アルゴリズムに渡される。古典最適化では、勾配降下法[13]、Nelder–Mead法[14]、COBYLA法[15]などを用いて、エネルギー期待値が小さくなるようにパラメータ $\theta$ を更新する。
UCCSD ansatzの場合、最適化されるパラメータは、単一励起および二重励起に対応する励起振幅である。すなわち、$t_i^a$ や $t_{ij}^{ab}$ に対応するパラメータを更新しながら、より低いエネルギーを与える試行波動関数を探索する。
4. 収束するまで反復する
VQEでは、量子コンピュータによる状態生成と測定、古典コンピュータによるパラメータ更新を繰り返す。エネルギー期待値 $E(\theta)$ が十分に収束したとき、その最小値を基底状態エネルギーの近似値とみなす。最適化されたパラメータは、
$$\theta^\ast=\arg\min_{\theta}\braket{\psi(\theta)|\hat{H}|\psi(\theta)}$$
と表される。このとき、得られる基底状態エネルギーの近似値は、
$$E_{\mathrm{VQE}}=\braket{\psi(\theta^\ast)|\hat{H}|\psi(\theta^\ast)}$$
である。変分原理により、この値は真の基底状態エネルギーに対する上界を与える。
結果
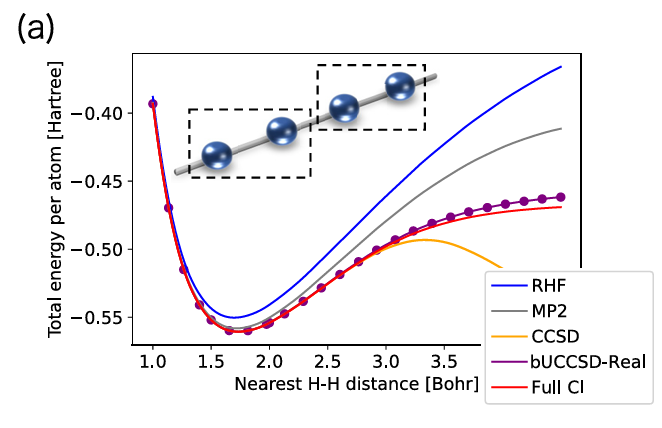
図1は、一次元水素原子鎖におけるポテンシャルエネルギー曲線を示している。横軸は隣接するH–H間距離、縦軸は原子当たりの全エネルギーである。結果に示されているbUCCSD-Real(broken-translational-symmetry UCCSD ansatz with real variables)は、通常のUCCSDを本論文で周期系材料に実装するために用いた変種である。通常のUCCSDは主に分子系を対象とするのに対し、bUCCSD-Real は並進対称性を厳密には課さず、周期系における電子相関をより柔軟に表現する。また、変分パラメータを実数に限定することで実装を簡略化している。量子回路の構成自体は通常の UCCSD とほぼ同様だが、対象とする系と対称性の扱いが異なる。
一次元水素鎖は、強相関電子系に対する電子状態計算手法の性能を評価するためのベンチマークとして用いられる。隣接するH–H間距離が大きくなると、電子の局在性が強まり、平均場的な近似では電子相関を十分に記述することが難しくなる。そのため、この領域ではFull CIの結果にどれだけ近いエネルギーを再現できるかが、計算手法の精度を評価する重要な指標となる。
図1の計算結果から、RHF法[16,17]、MP2法[18]、CCSD法[19]などの古典的な近似手法では、隣接H–H間距離が大きくなる強相関領域において、Full CIの結果から大きくずれることが分かる。一方、量子コンピュータ上での実装を想定したUCCSD型の手法であるbUCCSD-realは、H–H間距離が大きい領域でもFull CIに近いエネルギーを与えている。このことは、VQEに基づくUCCSD型ansatzが、強相関領域における電子相関を比較的よく記述できる可能性を示している。
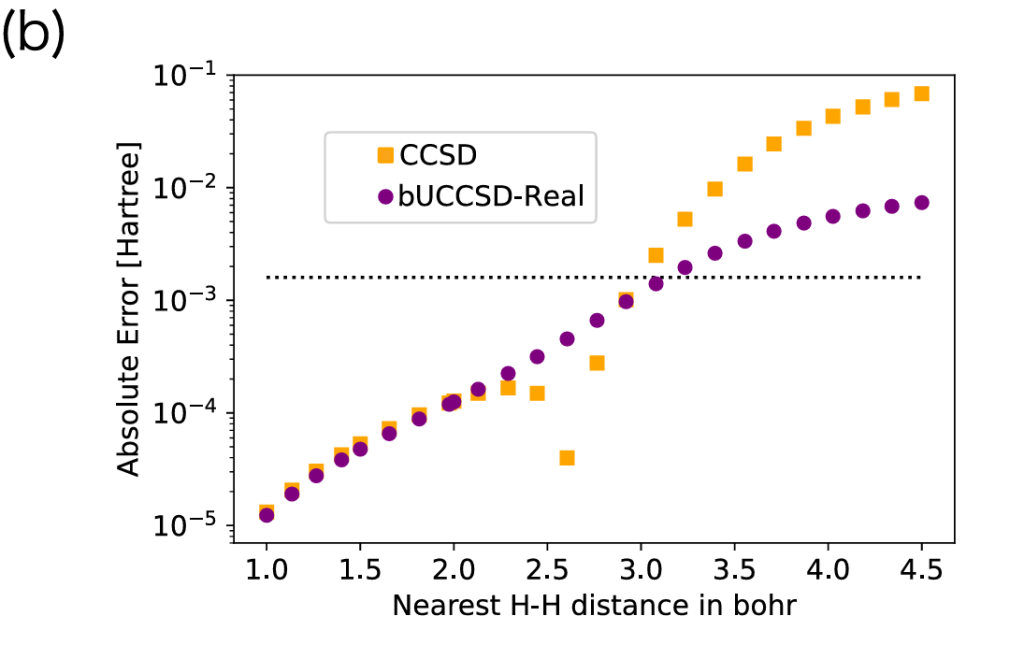
図2は、図1で示した各手法のうち、特にFull CIに近い結果を与えたCCSD法とbUCCSD-real法について、Full CIのエネルギーからの絶対誤差を比較したものである。横軸は隣接H–H間距離、縦軸はFull CIに対するエネルギーの絶対誤差である。
図2を見ると、H–H間距離が2.6〜2.8 Bohr付近では一時的にCCSD法の方が小さい誤差を示している。一方、3.0 Bohr以上の強相関領域では、bUCCSD-real法の方がFull CIに近いエネルギーを与えている。この結果から、bUCCSD-real法は、従来の古典的近似手法が苦手とする強相関領域において有効であることが示唆される。
まとめ
本記事では、周期的な一次元水素鎖に対するVQE計算を扱った研究について解説した。VQEは、量子コンピュータ上でパラメータ化された試行波動関数を生成し、そのエネルギー期待値を測定しながら、古典コンピュータ上でパラメータを最適化する量子–古典ハイブリッドアルゴリズムである。特にUCCSD型のansatzを用いることで、電子相関を取り入れた高精度な基底状態エネルギー計算が期待される。
論文で示された一次元水素鎖の計算結果では、隣接H–H間距離が大きくなり、電子相関の効果が強くなる領域において、RHF法、MP2法、CCSD法などの古典的近似手法ではFull CIからのずれが大きくなる。一方、VQEに基づくbUCCSD-real法は、強相関領域においてもFull CIに近いエネルギーを再現しており、周期系に対する量子計算手法の有効性を示している。
ただし、現実の量子デバイスでは、ノイズ、測定回数、量子ビット数、回路深さなどの制約が存在する。そのため、大規模な固体材料計算へ直接応用するには、まだ多くの課題が残されている。それでも、強相関電子系や従来手法で扱いにくい物質群に対して、VQEは将来的に有力な電子状態計算手法となる可能性がある。
このように、VQEは現時点では実機上の制約を受ける発展途上の手法であるものの、強相関電子系や周期系の電子状態計算に対する新しいアプローチとして重要な研究対象である。
参考文献
- Hafner, J., Wolverton, C., and Ceder, G., “Toward Computational Materials Design: The Impact of Density Functional Theory on Materials Research,” MRS Bulletin 31, 659–668 (2006).
- Hartree, D. R., “The Wave Mechanics of an Atom with a Non-Coulomb Central Field,” Mathematical Proceedings of the Cambridge Philosophical Society 24, 89–110 (1928).
- Fock, V., “Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems,” Zeitschrift für Physik 61, 126–148 (1930).
- Handy, N. C., “Multi-Root Configuration Interaction Calculations,” Chemical Physics Letters 74, 280–283 (1980).
- Kohn, W., and Sham, L. J., “Self-Consistent Equations Including Exchange and Correlation Effects,” Physical Review 140, A1133–A1138 (1965).
- Peruzzo, A., McClean, J., Shadbolt, P., Yung, M.-H., Zhou, X.-Q., Love, P. J., Aspuru-Guzik, A., and O’Brien, J. L., “A Variational Eigenvalue Solver on a Photonic Quantum Processor,” Nature Communications 5, 4213 (2014).
- Preskill, J., “Quantum Computing in the NISQ Era and Beyond,” Quantum 2, 79 (2018).
- Bharti, K., et al., “Noisy Intermediate-Scale Quantum Algorithms,” Reviews of Modern Physics 94, 015004 (2022).
- O’Malley, P. J. J., et al., “Scalable Quantum Simulation of Molecular Energies,” Physical Review X 6, 031007 (2016).
- Romero, J., Babbush, R., McClean, J. R., Hempel, C., Love, P. J., and Aspuru-Guzik, A., “Strategies for Quantum Computing Molecular Energies Using the Unitary Coupled Cluster Ansatz,” Quantum Science and Technology 4, 014008 (2018).
- Kandala, A., et al., “Hardware-Efficient Variational Quantum Eigensolver for Small Molecules and Quantum Magnets,” Nature 549, 242–246 (2017).
- Jordan, P., and Wigner, E., “Über das Paulische Äquivalenzverbot,” Zeitschrift für Physik 47, 631–651 (1928).
- Cauchy, A., “Méthode générale pour la résolution des systèmes d’équations simultanées,” Comptes Rendus de l’Académie des Sciences 25, 536–538 (1847).
- Nelder, J. A., and Mead, R., “A Simplex Method for Function Minimization,” The Computer Journal 7, 308–313 (1965).
- Powell, M. J. D., “A Direct Search Optimization Method That Models the Objective and Constraint Functions by Linear Interpolation,” in Advances in Optimization and Numerical Analysis, 51–67 (1994).
- Roothaan, C. C. J., “New Developments in Molecular Orbital Theory,” Reviews of Modern Physics 23, 69–89 (1951).
- Hall, G. G., “The Molecular Orbital Theory of Chemical Valency. VIII. A Method of Calculating Ionization Potentials,” Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences 205, 541–552 (1951).
- Møller, C., and Plesset, M. S., “Note on an Approximation Treatment for Many-Electron Systems,” Physical Review 46, 618–622 (1934).
- Purvis, G. D., and Bartlett, R. J., “A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples,” The Journal of Chemical Physics 76, 1910–1918 (1982).
Appendex A
結合クラスター法(Coupled Cluster: CC法)では、励起演算子 $T$ を用いて $\exp(T)$をHartree–Fock波動関数に作用させることで、Hartree–Fock法では記述できない電子相関を非線形に取り込む。例えば、励起演算子 $T$ として単一励起および二重励起のみを考慮したCCSD法であっても、$\exp(T)$の展開によって高次励起の効果を部分的に含むことができる。また、三重励起の寄与を摂動論的に取り入れたCCSD(T)法は、量子化学計算における標準的な手法として広く用いられている。
このようなCC法の考え方を量子コンピュータ上で利用できれば、高精度な電子状態計算への応用が期待される。しかし、通常のCC法で用いられる指数型クラスター演算子 $\exp(T)$ は一般にはユニタリー演算子ではないため、そのまま量子回路として実装することはできない。そこで、反エルミートな生成子 $T-T^\dagger$ を用いるユニタリー版のCoupled Cluster法、すなわちUnitary Coupled Cluster(UCC)法が量子計算用の波動関数ansatzとして提案・利用されている
結合クラスター法(Coupled Cluster: CC法)では、励起演算子 T を用いて、参照波動関数 $\ket{\Phi_0}$ に指数型演算子 \(e^T\) を作用させることで、電子相関を取り入れた波動関数を構成する。
$$\ket{\Psi_{\mathrm{CC}}}=e^T\ket{\Phi_0}$$
例えば、励起演算子 $T$ として単一励起および二重励起のみを考慮したCCSD法であっても、指数関数 $e^T$ の展開により、高次励起の効果を部分的に含むことができる。また、三重励起の寄与を摂動論的に取り入れたCCSD(T)法は、量子化学計算における高精度手法として広く用いられている。
一方、通常のCC法で用いられる $e^T$ は、一般にはユニタリー演算子ではない。そのため、そのまま量子回路として実装することは難しい。そこで、UCC法では反エルミート演算子 $T-T^\dagger$ を用いて、
$$\ket{\Psi_{\mathrm{UCC}}}=e^{T-T^\dagger}\ket{\Phi_0}$$
と波動関数を構成する。このとき、$T-T^\dagger$ は反エルミートであるため、$e^{T-T^\dagger}$ はユニタリー演算子となる。したがって、UCC法は量子コンピュータ上で実装しやすい波動関数ansatzとして利用されている。
Appendex B
UCCSD法の計算の詳細を載せる。
$T^{\dagger}$ は $T$ のエルミート共役であるから、$T$ が Hartree-Fock 波動関数における占有軌道から非占有軌道への電子励起を表すのに対して、$T^{\dagger}$ は非占有軌道から占有軌道への脱励起を表す。VQE による UCC 計算では$T$の式において二電子励起演算子までを含む UCCSD波動関数が用いられることが多い。ここで $|\Phi_0⟩$ が規格化されている場合、UCC エネルギーは以下のように表すことができる。
$$E_{\text{UCC}} = ⟨\Phi_0| \exp(\tau^\dagger)H \exp(\tau)\Phi_0⟩ = ⟨\Phi_0| \exp(−\tau)H \exp(\tau )|\Phi_0⟩$$
ここで $\tau^\dagger = (T − T^\dagger)^\dagger = T^\dagger − T = −\tau$ であることを用いた。この式は次式に示す Baker-Campbell-Housdorff の公式
$$\exp(−\tau)H \exp(\tau) = H +[H,\tau]+ \frac{1}{2!} [[H,\tau],\tau]+ \frac{1}{3!} [[[H,\tau],\tau],\tau] + \cdots$$
を用いて以下のように表される。
$$E_{UCC} =⟨\Phi_0|H|\Phi_0⟩+⟨\Phi_0|[H,τ]|\Phi_0⟩+ \frac{1}{
2!} ⟨\Phi_0|[[H,\tau],\tau]|\Phi_0⟩ + \cdots$$
ここで、普通の波動関数の時間発展演算子は$U =\exp(−iHt)$ の形になっているため例えばハミルトニアンの一電
子項を$h_{pq}(a^{\dagger}_{p}a_{q}+a^{\dagger}_{q}a_{p})$ という項として定義した。これは$a^{\dagger}_{p}a_q$ は複素関数であるが、$a^{\dagger}_{p}a_q+a^{\dagger}_qa_p$ という線形結合を取ることで虚部が消え、実部が残るからである。UCC波動関数において励起演算子は$\exp(\tau)=\exp(T −T^\dagger)$という形をしているため、$\tau$ は純虚数でなければならない。つまり、$a^{\dagger}_aa_i$ は実部を消すために$a^{\dagger}_aa_i−a^{\dagger}_ia_a$ という線形結合を取る必要がある。
UCC波動関数を生成するためには励起演算子の最適化が必要であり、そのためには古典コンピュータ上で励起振幅を最適化する必要がある。この最適化は古典コンピュータ上で行うため、VQEは古典コンピュータと量子コンピュータのハイブリッド型計算モデルであると言える。
一電子励起·脱励起演算子をJordan-Wigner変換を使ってパウリ演算子の積に変換すると以下のようになる。
$$t_{ia}(a^{\dagger}_aa_i − a^\dagger_ia_a) = \frac{i}{2} t_{ia}(X_a \otimes Z{a−1} \otimes \cdots \otimes Z_{i+1} \otimes Y_i −Y_a \otimes Z_{a−1} \otimes \cdots \otimes Z_{i+1} \otimes X_i)$$
UCC波動関数生成で参照波動関数に励起··· 脱励起演算子を作用させるときはクラスター演算子exp(T −T^{\dagger})をクラスター演算子の形で書き直すと以下のようになる。
$$\exp(t_{ia}(a^{\dagger}_aa_i − a^{\dagger}_ia_a)) = \exp( \frac{i}{2} t_{ia}(X_a \otimes Z{a−1} \otimes \cdots \otimes Z_{i+1} \otimes Y_i − Y_a \otimes Z_{a−1} \otimes \cdots \otimes Z_{i+1} \otimes X_i))$$
このようにしてUCC波動関数を生成することができる。
量子コンピュータ上でUCC波動関数が生成できた場合、UCC波動関数のエネルギー期待値を求める操作を行う。そのために第二量子化したハミルトニアンH を量子ビットハミルトニアンに書き直す必要がある。例としてH2分子の最小基底でのハミルトニアンをJordan-Wigner変換を用いて量子ビットハミルトニアンに書き直したものを以下に示す。
$$H=g_0+g_1(Z_1+Z_2)+g_2(Z_3+Z_4)+g_3Z_1Z_2+g_4Z_3Z_4+g_5(Z_1Z_3+Z_2Z_4)
+g_6(Z_1Z_4+Z_2Z_3)+g_7(X_1X_2Y_3Y_4+Y_1Y_2X_3X_4−X_1Y_2Y_3X_4−Y_1X_2X_3Y_4)$$
ここで$g_0∼g_7$は分子積分値から求まる係数である。この式に現れるパウリ演算子の添字は量子ビットのナンバリングであり、1番目、2番目の量子ビットはそれぞれ結合性分子軌道の$\alpha, \beta$スピン軌道、3番目、4番目の量子ビットはそれぞれ反結合性分子軌道の$\alpha, \beta$スピン軌道に対応している。ここでパウリ-Z演算子は$Z=\begin{pmatrix}
1 & 0 \\
0 &-1
\end{pmatrix}$として表されるので、例えば$g_1Z_1$は1番目の量子ビットが|0⟩のとき固有値$g_1$、|1⟩のとき固有値$−g_1$を返す。また、$g_3Z_1Z_2$は1番目と2番目の量子ビットの量子状態が|00⟩または|11⟩のとき固有値$g_3$、|01⟩または|10⟩のとき固有値$−g_3$を返す。一般にはパウリ-Z演算子の積の固有値は|1⟩状態にある量子ビットの数が偶数のときは+1、奇数のときは−1となる。そしてパウリ-X演算子とパウリ-Y演算子はそれぞれ$R_y(-\frac{π}{2}),R_x(\frac{π}{2})$ゲートを使ってX基底、Y基底をZ基底に変換し、対応する量子ビットの量子状態が|0⟩であるか|1⟩であるかによって判定する。
UCC法を用いたVQEではUCC波動関数の生成と測定を繰り返し、量子ビットハミルトニアンに含まれるパウリ演算子の積の期待値を求める。また、分子積分値をVQEの実行前に古典コンピュータで計算を行うことで求め、パウリ演算子の積の期待値に分子積分値から求めた係数をかけることで量子ビットハミルトニアンの各項の期待値が算出され、UCCエネルギーを計算できる。パウリ演算子の積の期待値を計算するためには測定を繰り返すことが必要だが、測定回数が少なければ統計誤差が大きくなる。しかし、無限回の測定を行うことはできないため、目安として量子ビットハミルトニアン各項にかかる係数の絶対値が最大のものを$w_{\text{max}}$とするとエネルギー計算値の誤差をε以下にするために$O(|w_{\text{max}}|^2/ε)$回程度の測定を繰り返すことが推奨される。
キーワードまとめ
- VQE VQE(Variational Quantum Eigensolver)は、量子コンピュータと古典コンピュータを組み合わせたハイブリッド型の変分量子アルゴリズムであり、量子系の基底状態エネルギーを近似的に求めることを目的とする。本手法では、パラメータ化された量子回路によって試行波動関数を生成し、その期待エネルギーを量子コンピュータ上で評価する。得られたエネルギー値に基づき、古典最適化アルゴリズムを用いて回路パラメータを反復的に更新することで、変分原理に従って基底状態へと収束させる。VQEは、比較的少ない量子ビット数および浅い量子回路で実装可能であるため、NISQ(Noisy Intermediate-Scale Quantum)デバイスに適した手法として広く研究されている。
- シュレディンガー方程式 シュレディンガー方程式は、量子力学における基本方程式であり、量子系の状態およびその時間発展を記述する。波動関数を未知関数とする偏微分方程式として定式化され、粒子の運動や相互作用を量子力学的に表現する基盤を与える。特に、時間に依存しないシュレディンガー方程式を解くことで、系の固有エネルギーおよび対応する固有状態を求めることができる。そのため、原子・分子・固体などの電子状態解析において中心的な役割を果たしている。
- 第一原理計算 第一原理計算(ab initio calculation)は、経験的パラメータを用いることなく、量子力学の基本法則に基づいて物質の電子状態や物性を予測する計算手法である。一般に、多電子系に対するシュレディンガー方程式を近似的に解くことで、原子配置や電子密度、全エネルギーなどを評価する。代表的な手法として密度汎関数理論(Density Functional Theory: DFT)が広く利用されている。第一原理計算は、材料設計、触媒開発、半導体研究など、多様な分野において重要な解析手法として位置付けられている。
- 量子化学計算 量子化学計算は、量子力学に基づいて分子系の電子状態を理論的に解析する計算手法の総称である。分子軌道法や配置間相互作用法、結合クラスター法などの手法を用いて、分子構造、反応エネルギー、電子分布および励起状態などを評価する。これにより、化学結合の形成機構や化学反応の進行過程を原子・電子レベルで理解することが可能となる。量子化学計算は、新規分子の設計、反応機構の解明、および材料・医薬品開発において重要な役割を担っている。
あとがき
この論文を読んで、量子コンピュータを活用した第一原理計算の研究がすでに進められていることを初めて知りました。本論文では、簡単な周期系を対象に、比較的高い精度と計算効率を両立した計算が実現されており、量子計算を物質科学へ応用する可能性を感じました。今後、表面や欠陥構造のようなより複雑な系に対しても、VQEを用いた計算の効率化が実現されることを期待しながら、この分野についてさらに勉強していきたいと思います。
東京科学大学物質理工学院材料系学士課程